Matthew T Sullenberger1*, Leanne H Kelley1*, and Eleanor M Maine1§
1Syracuse University
§Correspondence to: Eleanor M Maine (emmaine@syr.edu)
* Authors have equal contribution
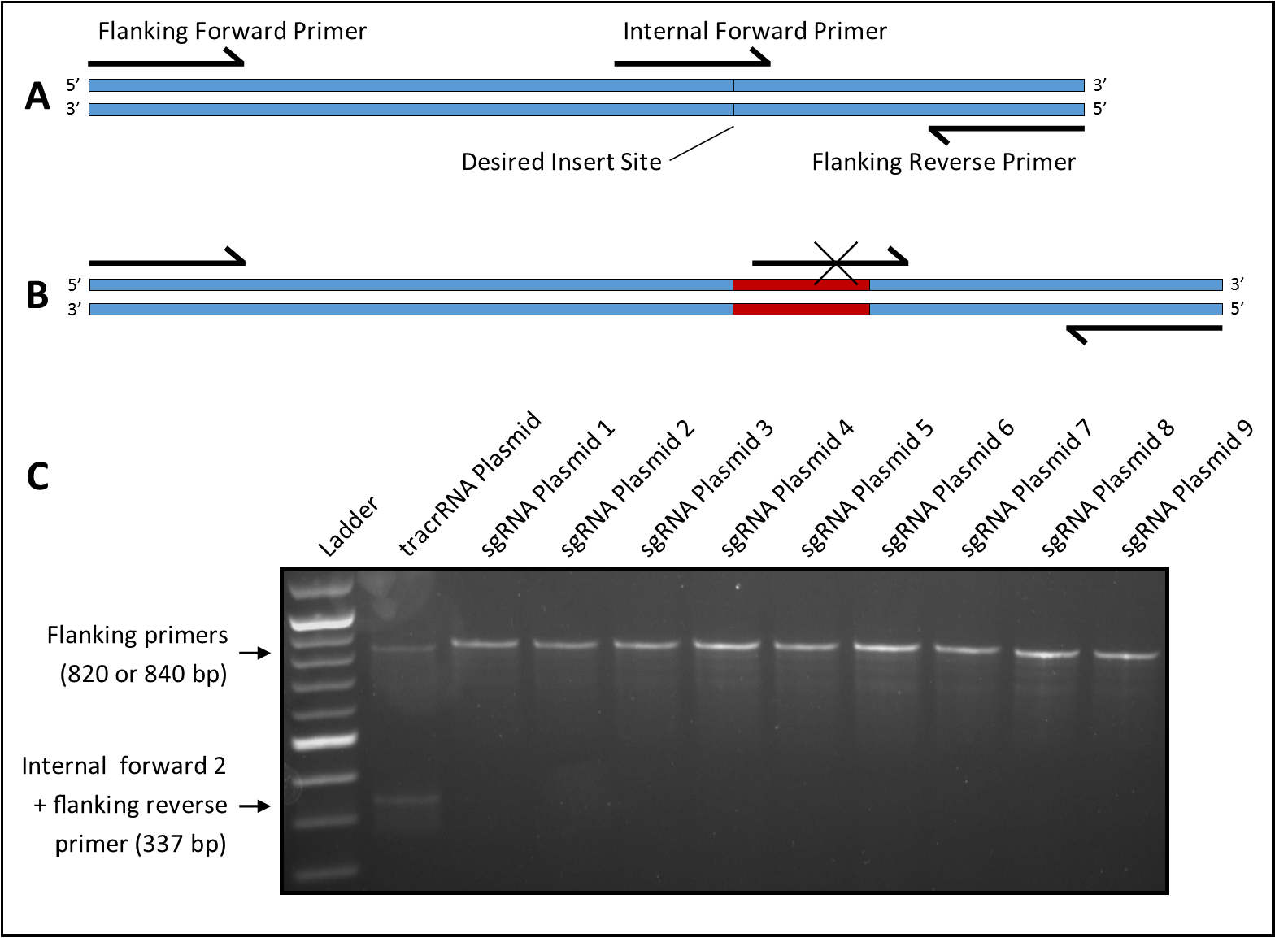
Description
Methods
Reagents
Funding
Author Contributions
- Matthew T Sullenberger: Conceptualization, Data curation, Formal analysis, Methodology, Visualization, Writing - original draft
- Leanne H Kelley: Conceptualization, Methodology, Writing - review and editing, Data curation, Formal analysis, Visualization
- Eleanor M Maine: Investigation, Funding acquisition, Project administration, Resources, Supervision, Writing - review and editing
Reviewed By
Jian Chen
History
- Received: 1/21/2020
- Accepted: 2/14/2020
- Published: 2/18/2020
Copyright
© 2020 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
PubMed Central: PMC7252386
PubMed: 32550507
microPublication Biology is published by
1200 E. California Blvd. MC 1-43 Pasadena, CA 91125
The microPublication project is supported by

The National Institute of Health -- Grant #: 1U01LM012672-01
microPublication Biology:ISSN: 2578-9430