Sonia El Mouridi1, Yuli Peng1, and Christian Frøkjær-Jensen1§
1King Abdullah University of Science and Technology (KAUST), Biological and Environmental Science and Engineering Division (BESE), KAUST Environmental Epigenetics Program (KEEP), Thuwal, 23955-6900, Saudi Arabia
§Correspondence to: Christian Frøkjær-Jensen (cfjensen@kaust.edu.sa)
Abstract
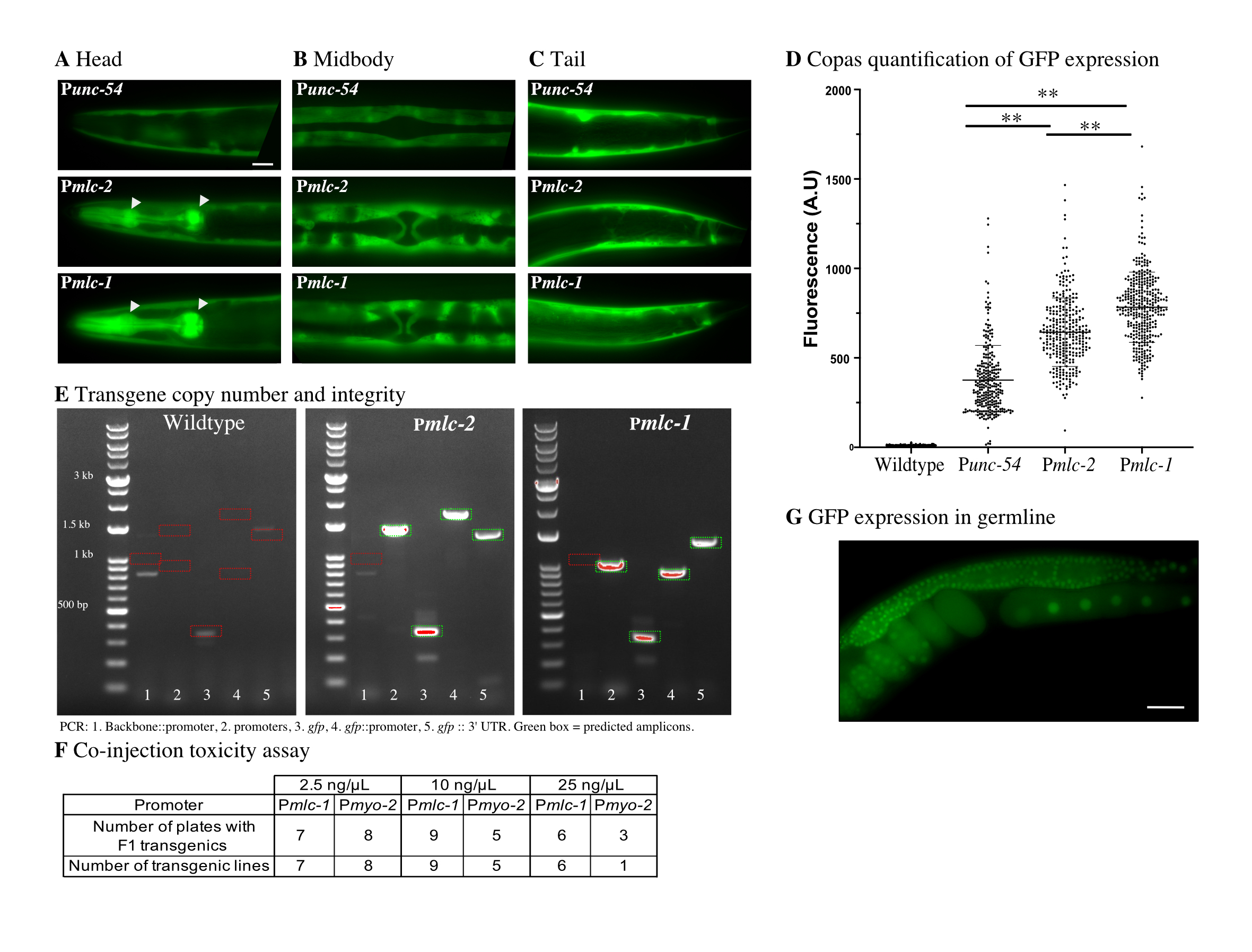
Description
Methods
Funding
Author Contributions
- Sonia El Mouridi: Investigation, Supervision, Writing - original draft
- Yuli Peng: Investigation, Writing - original draft
- Christian Frøkjær-Jensen: Funding acquisition, Conceptualization, Supervision, Writing - review and editing
Reviewed By
Daniel J Dickinson
Nomenclature Validated By
KJ Yook
Database Reference ID: WBPaper00060171
History
- Received: 8/24/2020
- Revision Received: 8/30/2020
- Accepted: 9/1/2020
- Published: 9/3/2020
Copyright
© 2020 by the authors. This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International (CC BY 4.0) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation
PubMed Central: PMC7474950
PubMed: 32908967
Cited By
microPublication Biology is published by
1200 E. California Blvd. MC 1-43 Pasadena, CA 91125
The microPublication project is supported by

The National Institute of Health -- Grant #: 1U01LM012672-01